Bravo API
1. Setup
The steps below describe how to setup the Bravo API on your server or local machine.
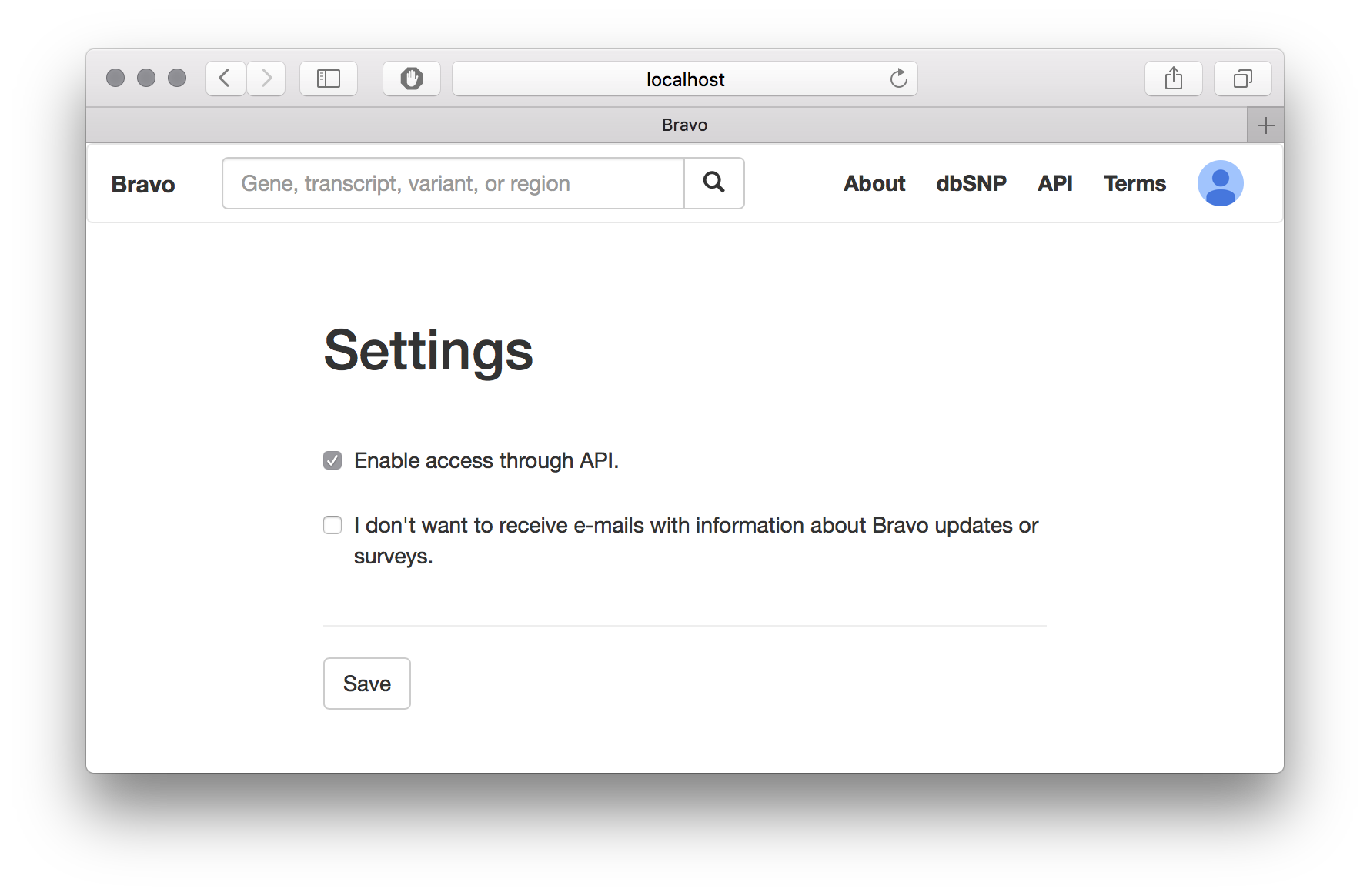
- Enable API access from your Profile->Settings page.
- Download the
bravocommand line tool for Linux and Mac OS X from here, or for Windows from here. Linux and Mac OS X users will need to set executable bits by runningchmod +x bravo. All code examples below are provided for Linux and Max OS X. - Run
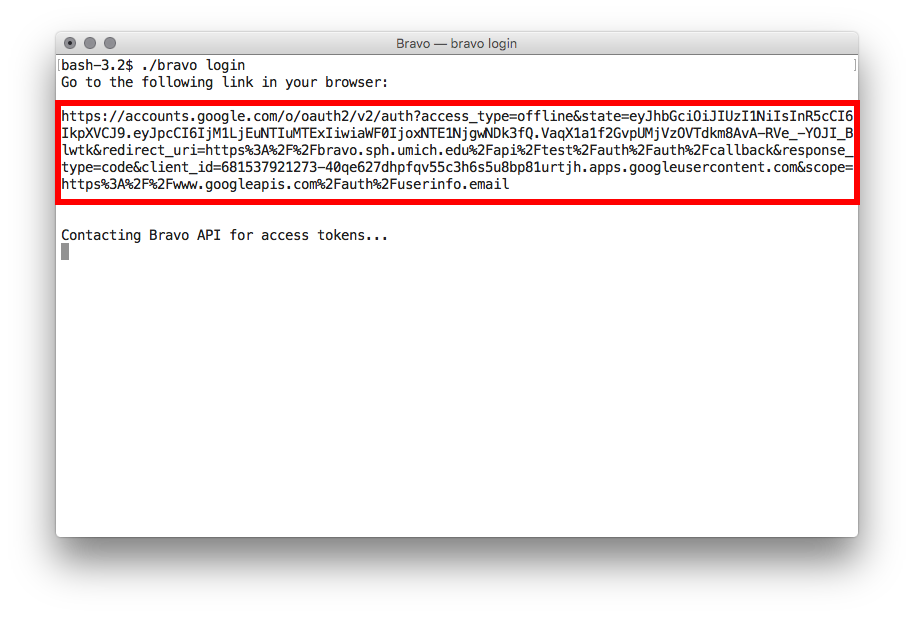
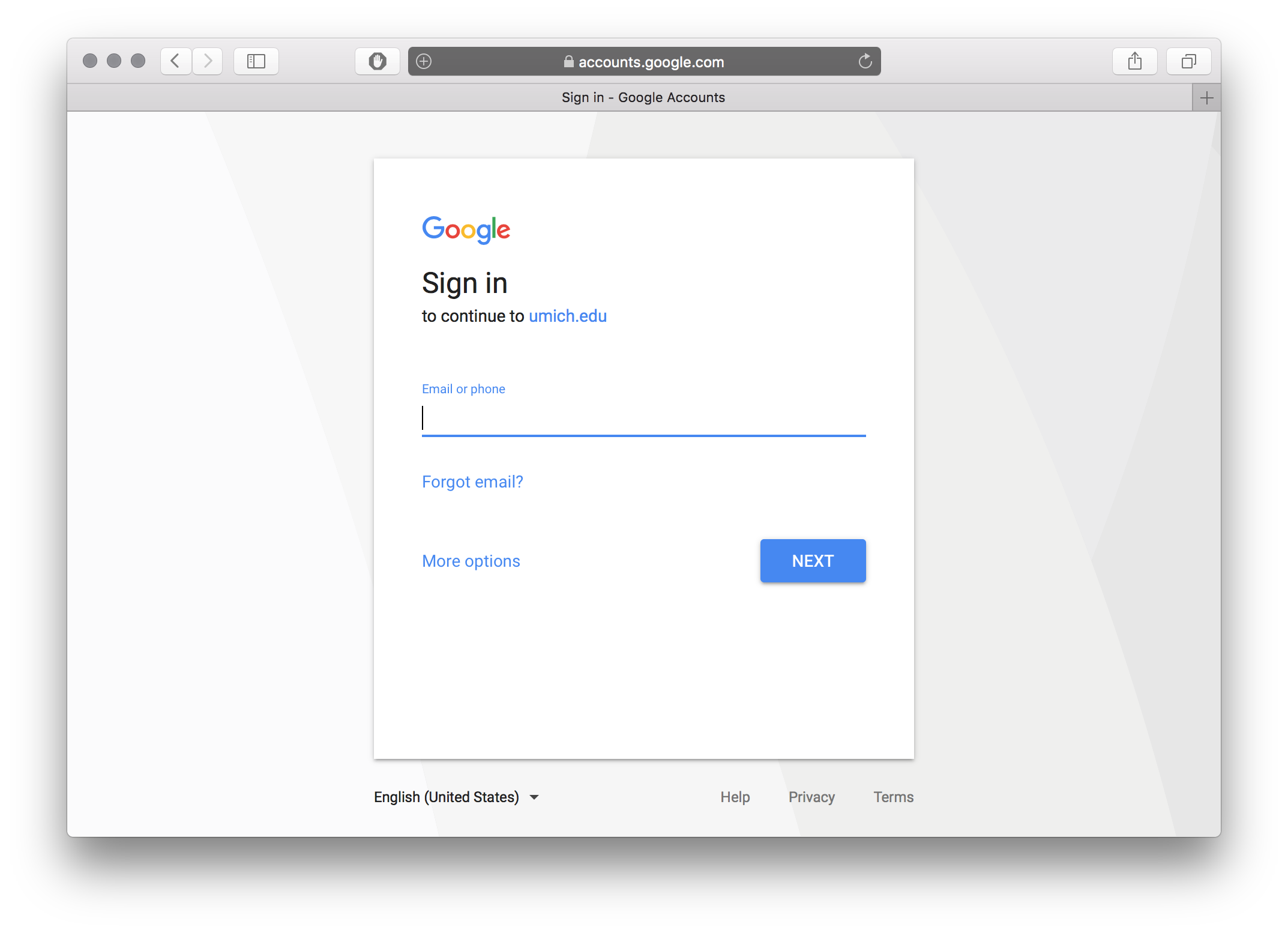
./bravo loginfrom your command line and go to the provided link in your browser. - Sign in with your Google account.
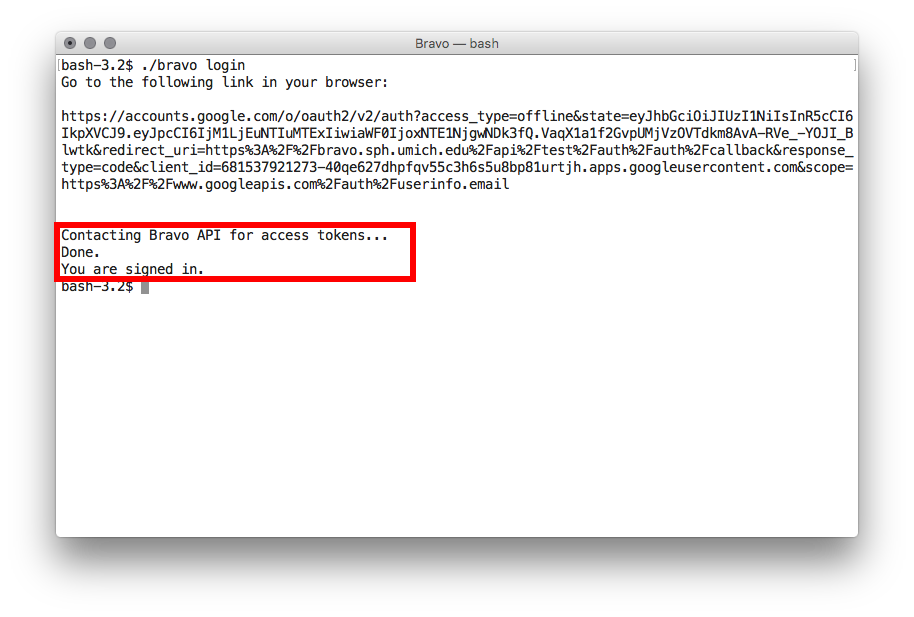
- Within several seconds
bravowill receive your authorization and you may start using the Bravo API. - To get your access token, execute
./bravo print-access-token. - Each request to the Bravo API must have
Authorizationheader with a valid Bearer access token. For example, to make an API call withcurl, execute:
curl -H "Authorization: Bearer `./bravo print-access-token`" https://bravo.sph.umich.edu/freeze5/hg38/api/v1/ - You need to login using
./bravo logineach time you access the Bravo API from within a different network.
2. Constraints
- Your Bravo API access token is tied to your public IP address.
- You can submit up to 180 queries within a 15 minute window. When this threshold is exceeded, queries will not be processed until the 15 minute window passes.
- All query results are paginated with a maximum of 1,000 variants per page.
- Region queries are restricted to up to 250,000 base-pairs.
3. Responses
The Bravo API sends a response as a JSON object with three mandatory keys data, format and next.
The data key points to the array of variants that satisfies your query, format specifies the format in which the varints are stored in data, and next stores the link to the next results page.
{
"data": [ ... ],
"format": "...",
"next": "https://bravo.sph.umich.edu/..."
}
3.1. JSON
By default, variants in the data array are represented as JSON objects.
{
"data": [
{
"allele_count": 2,
"allele_freq": 1.59276e-05,
"allele_num": 125568,
"alt": "G",
"chrom": "22",
"filter": "PASS",
"pos": 16387678,
"ref": "C",
"site_quality": 255.0,
"variant_id": "22-16387678-C-G"
},
{
"allele_count": 1,
"allele_freq": 7.96381e-06,
"allele_num": 125568,
"alt": "T",
"chrom": "22",
"filter": "PASS",
"pos": 16387678,
"ref": "C",
"site_quality": 157.0,
"variant_id": "22-16387678-C-T"
}
],
"format": "json",
"next": null
}
3.2. VCF
To encode variants inside the data array in VCF format, you must specify the vcf=1 (or vcf=True) option as a parameter in your GET query.
When using VCF format, additional header and meta keys in the response JSON object store the VCF header line and VCF meta-information lines, respectively.
{
"data": [
"22\t16387678\t22-16387678-C-G\tC\tG\t255.0\tPASS\tAN=125568;AC=2;AF=1.59276e-05",
"22\t16387678\t22-16387678-C-T\tC\tT\t157.0\tPASS\tAN=125568;AC=1;AF=7.96381e-06"
],
"format": "vcf",
"header": "#CHROM\tPOS\tID\tREF\tALT\tQUAL\tFILTER\tINFO",
"meta": [
"##fileformat=VCFv4.2",
"##FILTER=<ID=PASS,Description=\"All filters passed\">",
"##FILTER=<ID=CEN,Description=\"Variant located in centromeric region with inferred sequences\">",
"##FILTER=<ID=SVM,Description=\"Variant failed SVM filter\">",
"##FILTER=<ID=DISC,Description=\"Mendelian or duplicate genotype discordance is high (3/5% or more)\">",
"##FILTER=<ID=CHRXHET,Description=\"Excess heterozygosity in chrX in males\">",
"##FILTER=<ID=EXHET,Description=\"Excess heterozygosity with HWE p-value < 1e-6\">",
"##INFO=<ID=AN,Number=1,Type=Integer,Description=\"Number of Alleles in Samples with Coverage\">",
"##INFO=<ID=AC,Number=A,Type=Integer,Description=\"Alternate Allele Counts in Samples with Coverage\">",
"##INFO=<ID=AF,Number=A,Type=Float,Description=\"Alternate Allele Frequencies\""
],
"next": null
}
3.3. Pagination
The Bravo API splits large query results into pages of up to 1,000 variants.
A link to the next page is stored in the next key in the response JSON object.
If no more results are available, then next will have null value.
You may change the number of variants returned per page with the limit=N query parameter, where N is any integer between 1 and 1,000.
3.4. Handling errors
Upon success, the Bravo API sends the response with HTTP status code 200.
If client error, the response has HTTP status code 400, and the returned JSON object has only one key, error, with error message.
4. Queries
The following table lists all Bravo API endpoints currently available at https://bravo.sph.umich.edu/freeze5/hg38/api/v1.
| API endpoint | Description |
|---|---|
/ |
Retrieve meta-information about dataset. |
/variant |
Query single variant by chromosomal position or identifier. |
/region |
Query all variants within a chromosomal region. |
/gene |
Query all variants within a gene. |
4.1. Single variant query
To query a single variant, the GET request must be sent to https://bravo.sph.umich.edu/freeze5/hg38/api/v1/variant.
The following table lists all supported parameters.
| Parameter | Required | Description |
|---|---|---|
variant_id |
Only if single parameter | Variant identifier CHROM-POS-REF-ALT. |
chrom |
Only with pos |
Chromosome name. |
pos |
Only with chrom |
Chromosomal position in base-pairs. |
vcf |
No | Format output as VCF entries. Default is 0 (False). |
4.1.1. Examples
- Find a variant with chr22-16389447-A-G identifier:
curl -H "Authorization: Bearer `./bravo print-access-token`" "https://bravo.sph.umich.edu/freeze5/hg38/api/v1/variant?variant_id=chr22-16389447-A-G".{ "data": [ { "allele_count": 6261, "allele_freq": 0.0498614, "allele_num": 125568, "alt": "G", "chrom": "22", "filter": "PASS", "pos": 16389447, "ref": "A", "site_quality": 255.0, "variant_id": "22-16389447-A-G" } ], "format": "json", "next": null } - Find all variants at position 16,390,137 bp on chromosome 22:
curl -H "Authorization: Bearer `./bravo print-access-token`" "https://bravo.sph.umich.edu/freeze5/hg38/api/v1/variant?chrom=chr22&pos=16390137".{ "data": [ { "allele_count": 2, "allele_freq": 1.59276e-05, "allele_num": 125568, "alt": "A", "chrom": "22", "filter": "PASS", "pos": 16390137, "ref": "T", "site_quality": 255.0, "variant_id": "22-16390137-T-A" }, { "allele_count": 1, "allele_freq": 7.96381e-06, "allele_num": 125568, "alt": "C", "chrom": "22", "filter": "PASS", "pos": 16390137, "ref": "T", "site_quality": 255.0, "variant_id": "22-16390137-T-C" } ], "format": "json", "next": null }
3.2. Region query
To query all variants within a region, the GET request must be sent to https://bravo.sph.umich.edu/freeze5/hg38/api/v1/region.
The following table lists all supported parameters.
| Parameter | Required | Description |
|---|---|---|
chrom |
Yes | Chromosome name. |
start |
Yes | Chromosomal start position in base-pairs. |
end |
Yes | Chromosomal end position in base-pairs. |
allele_count |
No | Filter variants by alternate allele count in samples with coverage. |
allele_freq |
No | Filter variants by alternate allele frequency. |
allele_num |
No | Filter variants by number of alleles in samples with coverage. |
site_quality |
No | Filter variants by phred-scaled quality score. |
filter |
No | Filter variants by their status, e.g. PASS. |
sort |
No |
Comma-separated list of fields on which to sort the results.
Sort directions asc (ascending) or desc (descending) can optionally be appended to each field separated by the : character.
The following fields can be sorted: pos, allele_count, allele_freq, allele_num, site_quality, filter.
By default, results are sorted by chromosome position in ascending order.
|
limit |
No | Number of variants per results page. Default is 1,000. |
vcf |
No | Format output as VCF entries. Default is 0 (False). |
3.2.1. Filtering
Bravo API supports following comparison operators: eq (equal), ne (not equal), gt (greater than), lt (less than), gte (greater than or equal to), lte (less than or equal to).
Operators must be followed by the : character, so the query allele_freq=gte:0.01 searches for variants with allele frequency less than or equal to 0.01.
Simple equality is the default operation, and is performed as filter=PASS (same as filter=eq:PASS).
If needed, you can escape operators with quotes, so the query filter="ne:" searches for variants with "ne:" in the filter field.
3.2.2. Examples
- Extract all non-PASS variants between 16,387,675 bp and 16,390,908 bp on chromosome 22, sort results by decreasing allele frequency, and paginate result into pages of up to 5 variants:
curl -H "Authorization: Bearer `./bravo print-access-token`" "https://bravo.sph.umich.edu/freeze5/hg38/api/v1/region?chrom=chr22&end=16390908&start=16387675&filter=ne:PASS&sort=allele_freq:desc&limit=5{ "data": [ { "allele_count": 3392, "allele_freq": 0.0270133, "allele_num": 125568, "alt": "C", "chrom": "22", "filter": "SVM;DISC;EXHET", "pos": 16390907, "ref": "T", "site_quality": 143.0, "variant_id": "22-16390907-T-C" }, { "allele_count": 2610, "allele_freq": 0.0207856, "allele_num": 125568, "alt": "C", "chrom": "22", "filter": "SVM;DISC;EXHET", "pos": 16390901, "ref": "A", "site_quality": 117.0, "variant_id": "22-16390901-A-C" }, { "allele_count": 150, "allele_freq": 0.00119457, "allele_num": 125568, "alt": "C", "chrom": "22", "filter": "SVM;DISC", "pos": 16389436, "ref": "G", "site_quality": 27.0, "variant_id": "22-16389436-G-C" }, { "allele_count": 21, "allele_freq": 0.00016724, "allele_num": 125568, "alt": "AAGCAGCGGCTGG", "chrom": "22", "filter": "SVM", "pos": 16390735, "ref": "A", "site_quality": 147.0, "variant_id": "22-16390735-A-AAGCAGCGGCTGG" }, { "allele_count": 11, "allele_freq": 8.76019e-05, "allele_num": 125568, "alt": "A", "chrom": "22", "filter": "SVM", "pos": 16390780, "ref": "C", "site_quality": 255.0, "variant_id": "22-16390780-C-A" } ], "format": "json", "next": "https://bravo.sph.umich.edu/freeze5/hg38/api/v1/region?sort=allele_freq:desc&filter=ne:PASS&end=16390908&start=16387675&limit=5&chrom=chr22&last=59a8c9b38e3f1b1a035a5475:8.76019e-05" }
3.2. Gene query
In addition to region query which allows extracting variants between explicitely specified start and end chromosomal positions, the Bravo API allows you to query variants within a specified gene.
Given a gene name or gene identifier, the Bravo API will lookup corresponding chromosome and gene boundaries for you.
To query all variants within a region, the GET request must be sent to https://bravo.sph.umich.edu/freeze5/hg38/api/v1/gene.
The following table lists all supported parameters.
| Parameter | Required | Description |
|---|---|---|
name |
Yes | Gene name or gene identifier. | allele_count |
No | Filter variants by alternate allele count in samples with coverage. |
allele_freq |
No | Filter variants by alternate allele frequency. |
allele_num |
No | Filter variants by number of alleles in samples with coverage. |
site_quality |
No | Filter variants by phred-scaled quality score. |
filter |
No | Filter variants by their status, e.g. PASS. |
sort |
No |
Comma-separated list of fields on which to sort the results.
Sort directions asc (ascending) or desc (descending) can optionally be appended to each field separated by the : character.
The following fields can be sorted: pos, allele_count, allele_freq, allele_num, site_quality, filter.
By default, results are sorted by chromosome position in ascending order.
|
limit |
No | Number of variants per results page. Default is 1,000. |
vcf |
No | Format output as VCF entries. Default is 0 (False). |
3.2.1. Examples
- Extract non-PASS variants in gene ABCD1P4 sorted by alternate allele frequency in increasing order, receive 5 variants at a time:
curl -H "Authorization: Bearer `./bravo print-access-token`" "https://bravo.sph.umich.edu/freeze5/hg38/api/v1/gene?name=ABCD1P4&filter=ne:PASS&sort=allele_freq:asc&limit=5"{ "data": [ { "allele_count": 1, "allele_freq": 7.96381e-06, "allele_num": 125568, "alt": "C", "chrom": "22", "filter": "SVM", "pos": 16387844, "ref": "G", "site_quality": 50.0, "variant_id": "22-16387844-G-C" }, { "allele_count": 1, "allele_freq": 7.96381e-06, "allele_num": 125568, "alt": "T", "chrom": "22", "filter": "SVM", "pos": 16388018, "ref": "C", "site_quality": 44.0, "variant_id": "22-16388018-C-T" }, { "allele_count": 1, "allele_freq": 7.96381e-06, "allele_num": 125568, "alt": "A", "chrom": "22", "filter": "SVM", "pos": 16388182, "ref": "C", "site_quality": 32.0, "variant_id": "22-16388182-C-A" }, { "allele_count": 1, "allele_freq": 7.96381e-06, "allele_num": 125568, "alt": "C", "chrom": "22", "filter": "SVM", "pos": 16388371, "ref": "T", "site_quality": 255.0, "variant_id": "22-16388371-T-C" }, { "allele_count": 1, "allele_freq": 7.96381e-06, "allele_num": 125568, "alt": "T", "chrom": "22", "filter": "SVM", "pos": 16388398, "ref": "G", "site_quality": 255.0, "variant_id": "22-16388398-G-T" } ], "format": "json", "gene": { "chrom": "22", "id": "ENSG00000225293", "name": "ABCD1P4", "start": 16387695, "stop": 16390888, "strand": "+" }, "next": "https://bravo.sph.umich.edu/freeze5/hg38/api/v1/gene?filter=ne:PASS&sort=allele_freq:asc&limit=5&name=ABCD1P4&last=59a8c9b38e3f1b1a035a516b:7.96381e-06" }
5. Tutorial
The Bravo API tutorial with step-by-step examples is available from here.